Resümee BBS Talk 2
Amyloid - Targets einer kausalen Alzheimer-Therapie
Professor Doktor Richard Dodel, Neurologie Chefarzt und Inhaber eines Lehrstuhls für Geriatrie an der Universität Duisburg Essen, gibt einen Überblick über die therapeutischen Optionen einer kausalen Alzheimer-Therapie, wobei der Fokus auf Amyloid als Targets liegt [1].
Redaktion: Prime Public Media (PPM), Zürich
Im Mittelpunkt der Alzheimer Erkrankung stehen nach wie vor zwei Veränderungen: Die Neurofibrillären Tangles und die Amyloid-Plaques, die bereits 1907 von Alois Alzheimer beschrieben wurden. Die Amyloid-Plaques entstehen aus einem grösseren Amyloid Präkursor Protein. Dabei ist das Abeta in die Membran eingelagert und wird von der Beta- und Gamma-Sekretase gespalten, wobei Abeta freigesetzt wird. Wird darüber hinaus auch die Alpha-Sekretase tätig, entsteht ein sogenanntes Peptide p3. Daraus abgeleitete Therapieansätze zur Behandlung der alzheimerischen Erkrankung, wie die Hemmung der Beta- und Gamma-Sekretase und die Aktivierung der Alpha-Sekretase sind dennoch leider nicht zur therapeutischen Reife gelangt.
Der Amyloid Kaskadenhypothese zufolge ist die Bildung von Abeta 42 durch nacheinander folgende Schritte der Beta-Sekretase und der Gamma-Sekretase im APP-Molekül Auslöser der Alzheimer-Problematik. Im Mittelpunkt steht erhöhtes Amyloid-Beta das einerseits durch eine erhöhte Produktion oder einen erniedrigten Metabolismus ansteigt. Extrazellulär bilden Abeta 42-Moleküle zunächst kleinere Aggregate bevor sie sich zu grossen Aggregaten, den Amyloid- Plaques, zusammenlagern. Darüber hinaus kommt es zu Veränderungen in den Neurofibrillen, welche das Tau- Protein hyperphosphorylieren.
Aktualisiertes hypothetisches Modell dynamischer Biomarker
Ein hypothetisches Modell der wichtigsten Biomarker der Alzheimer-Krankheit (AD) zeigt die zeitliche Entwicklung von AD-Biomarkern im Verhältnis zueinander und zum Auftreten und Fortschreiten klinischer Symptome. Abbildung 1 zeigt in hellgrün dargestellt das Stadium der Demenz sowie das MCI-Stadion und die normale Kognition. Mittlerweile gibt es Biomarker, die Jahre bis Jahrzehnte vor Beginn der Erkrankung positiv werden, dargestellt durch die Linien auf der linken Seite. Das Amyloid-Beta steht dabei ganz am Anfang der Kaskade, gefolgt von der Tau-Aktivierung (Abb. 1) [2]. Eine Pipeline-Analyse von Medikamenten in klinischen Studien zur Behandlung von AD zeigt zudem, dass biologische Zielprozesse stärker diversifiziert sind, Biomarker regelmässiger verwendet werden und wiederverwendete Wirkstoffe untersucht werden, um ihren Nutzen für die Behandlung von AD zu bestimmen. Wobei die meisten Wirkstoffe in der Studie auf eine Krankheitsmodifikation abzielen (Abb. 2) [3].
Die Passive Immunisierung
Zu den wichtigsten Amyloid-senkenden Therapieansätzen gehören neben der Hemmung der Beta-Amyloid-Produktion durch Beta- und Gamma-Sekretasehemmer und der Hemmung der Beta-Amyloid-Aggregation durch Metallchelatoren, der Abbau von Beta-Amyloid aus dem Gehirn durch eine entsprechende Immunisierung. Für die passive Immunisierung stehen verschiedene Antikörper zur Verfügung. Hierzu zählen die vollständig humanen Antikörper, die humanisierten Antikörper sowie einzelne Fragmente eines Antikörpers, welche überwiegend im universitären Umfeld verwendet werden und die Nanobodies. Die häufigsten Antikörper sind gegen körperfremde Antigene gerichtet. Natürlich vorkommenden Antikörper können sich hingegen gegen körperfremde und körpereigene Antigene richten. Der Unterschied besteht darin, dass natürlich vorkommenden Antikörper definiert sind, während die häufigen auftretenden Antikörper kombinatorisch aus verschiedenen Ketten entstehen können. Durch die Weiterentwicklung der Antikörper sind mittlerweile sogenannte VH Gene entstanden. Diese Antikörper sind sowohl gegen körpereigene als auch gegen veränderte körpereigene Komponenten gerichtet. Sie sind meist polyklonal und stellen die erste Immunabwehr gegen Viren und Bakterien dar. Zwei Drittel des menschlichen Antikörper Pools bestehen aus diesen natürlich vorkommenden Autoantikörpern, die an Proteine und DNA binden können. Das bekannteste System ist das ABO-System, das gewebshomöostatisch eine wichtige Rolle spielt [4,5].
Im Verlauf der Immunantwort erfüllen Antikörper verschiedene Funktionen. Sie dienen zum einen dazu eingedrungene Antigene abzufangen und zu blockieren, so dass sie ihre schädliche Wirkung nicht entfalten können, oder es wird verhindert, dass das Antigen mit Körperzellen interagiert. Das Epitop ist der Teil eines Antigens, das von dem Antikörper erkannt wird. Auf Abeta befindet sich beispielsweise ein B-Zell Epitop, welches von den B-Zellen erkannt wird; ein Epitop für die Abeta-Effektivität, sprich für die Phagocytose des Amyloids sowie ein Epitop für die T-Zell Aktivierung. Die meisten Abeta-Antikörper richten sich gegen N-terminale Regionen.
Die neuen Targets für Antikörper sind eher Dimere oder Aggregate als Monomere, wie es bei den neueren Substanzen (Aducanumab, Gantenerumab, Lecanemab und Donanemab) der Fall ist. Das liegt an der Abeta-Oligomerisierung. Das Amyloid bildet N-Terminal zu C-Terminal eine Haarnadel-Schleife, die aggregiert. Durch diese hauptsächlich über den C-Terminus gebildete Aggregation entsteht ein neues Epitop, welches im Gegensatz zu dem alten Epitop deutlich an dem Endstück herausragt und somit leichter angegriffen werden kann. Amyloid-Beta-Protein-Dimere, die direkt aus Alzheimer-Gehirnen isoliert wurden, beeinträchtigen die synaptische Plastizität und das Gedächtnis [6].
Amyloid-Plaque Veränderungen in klinischen Studien
Studien mit Donanemab, Aducanumab und Lecanemab haben in Phase zwei eine deutliche Reduktion der Amyloid-Plaques gezeigt. Was darauf schliessen lässt, dass zwischen der Abräumung der Amyloid-Plaques und der Kognition ein klarer Zusammenhang besteht. Dieser wurde in Studien mit Aducanumab und monoklonalen Anti-Abeta-Antikörpern bestätigt, bei denen ein grösserer Behandlungseffekt auf die Abeta-Plaques im Gehirn mit einem grösseren Behandlungseffekt auf CDR-SB in Verbindung gebracht werden konnte [7]. In den ersten Immunisierungsstudien konnte zwar gezeigt werden, dass Plaques abgeräumt werden können, sich die Kognition aber nicht verbessert. Diese Veränderung in den klinischen Studien ist darauf zurückzuführen, dass Therapien heutzutag viel früher an Patienten herangetragen werden können. Der entscheidende Wirkmechanismus dabei ist nicht der Abbau von Plaques, sondern der Aufbau dieser. Die potentiell beste Therapiezeit für den Eingriff in den Pathomechanismus besteht, wenn Abeta bereits positiv im Liquor ist, nicht aber Tau. Hierfür kommen zwei Therapieansätze in Frage, der periphere und der zentrale Abbau. Beim zentralen Abbau greifen Antikörper die Plaques im Gehirn an. Die Glia-Zellen bauen dann den Antikörper und das gebundene Abeta ab. Beim peripheren Abbau hingegen verbleiben die Antikörper im Blut, durch einen Austausch ist der Abbau von Abeta auch hier möglich. Zurzeit kann nicht mit Sicherheit bestimmt werden, welcher Abbau wann eintritt. Um den Wirkmechanismus genauer bestimmen zu können, müssen die unterschiedlichen Antikörper weiter charakterisiert werden [8].
Abbildung 1: Überarbeitete dynamische Biomarker des pathologischen Kaskadenmodells der Alzheimer-Krankheit - 2012
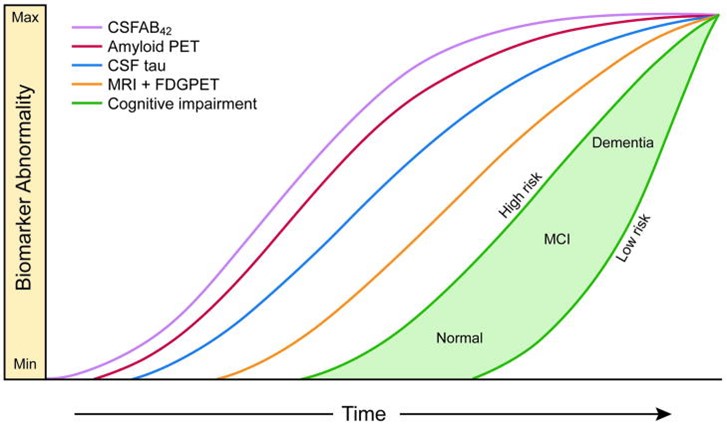
Quelle nach [2].
Abbildung 2: Wirkstoffe in klinischen Studien zur Behandlung der Alzheimer-Krankheit im Jahr 2021
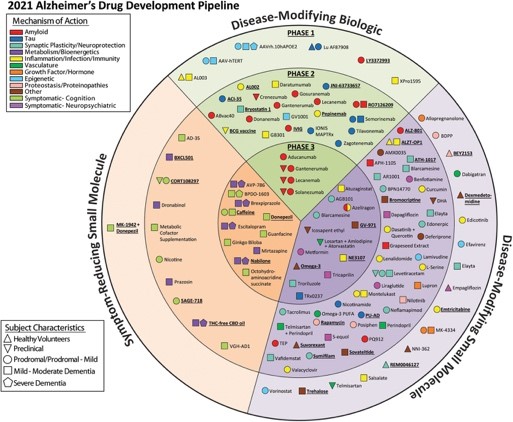
Der innere Ring zeigt Phase-3-Agenten; der mittlere Ring enthält Phase-2-Agenten; der äußere Ring präsentiert Therapien der Phase 1; Wirkstoffe in Grünflächen sind Biologika; violette Wirkstoffe sind krankheitsmodifizierende kleine Moleküle; Wirkstoffe in Orange sind symptomatische Wirkstoffe, die sich mit kognitiver Verbesserung oder Verhaltens- und neuropsychiatrischen Symptomen befassen; die Form des Symbols zeigt die Population der Studie; die Symbolfarbe zeigt die auf der Common Alzheimer's Disease Research Ontology (CADRO) basierende Klasse des Wirkstoffs (die Kategorie „Sonstige“ umfasst CADRO-Klassen mit drei oder weniger Wirkstoffen in Studien). Unterstrichene Agenten sind seit 2020 neu in der Pipeline. Abbildung: J Cummings; M de la Flor, PhD,Illustrator
Quelle nach [3].
Literatur:
1. Dodel, Richard: Amyloid als Targets einer kausalen Alzheimer-Therapie. BBS Talk zu dem Thema „Update Demenz“ 2021.
2. Jack et al.: Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013, doi: 10.1016/S1474-4422(12)70291-0.
3. Cummings et al.: Alzheimer's disease drug development pipeline: 2021. Alheimer’s Dement 2021, doi: https://doi.org/10.1002/trc2.12179.
4. Schoenfeld et al.: Active immunotherapy induces antibody responses that target tumor angiogenesis. Cancer Res 2010, doi: 10.1158/0008-5472.CAN-10-1852.
5. Xu et al.: A recurring motif for antibody recognition of the receptor-binding site of influenza hemagglutinin. Nat Struct Mol Biol 2013, doi: 10.1038/nsmb.2500.
6. Shankar et al.: Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat med 2008, doi: 10.1038/nm1782.
7. Rajagovindan et al.: Abeta, amyloid beta; CDR-SB, Clinical Dementia Rating Scale-Sum of Boxes; PET, poitron emission tomography; SUVR, standardized uptake value ratio. Data presented at AAIC 2021.
8. Dodel et al.: Immunotherapy for Alzheimer's disease. Lancet Neurol 2003, doi: 10.1016/s1474-4422(03)00349-1.